

The medical device industry in India is fairly new. According to Infosys, around 75 per cent of the medical equipment is imported. And the equipment is not always suitable for Indian requirements.
Why are the medical devices not being made sufficiently in India? There are some unique challenges to be addressed here. Let us take a look.
Challenges involved in medical device design
The first problem that medical device designers encounter is that, they need to have interdisciplinary knowledge to be able to design good-quality products. It is not sufficient to have a good medical background, because every consumer medical product also has a substantial electronic component that needs to be designed. Thus, there needs to be a collaboration between the doctors and engineers to ensure that the product gives a good overall performance. This collaboration is challenging, because engineers and doctors are not usually trained to work together.



The other problem is that, the medical device market is highly regulated. No hospital would purchase a medical product unless authorities approve it. Thus, the product has to meet all requirements of the regulatory authorities before it can enter the market.
We do not have a regulation set-up in India yet for medical regulation bodies. A bill proposed to solve this is due in the Parliament for approval. Central Drugs Standard Control Organization (CDSCO) is responsible for some level of device regulation in India.
There is also a new amendment in discussion with respect to existing rules, for which a draft has been introduced for public comments. Known as Medical Device Rules, 2016, it suggests categorisation of medical devices into four groups, depending on the risks associated with the device.
Dealing with the challenges
While designing a new medical device, it is important to do some research and consult with multiple doctors. This is called the process of concept validation. Information from doctors is very helpful. At this stage, they can provide feedback regarding clinical issues that the concept may have. The design can be improved to overcome these issues.
After the doctors approve the concept, the real design process begins, involving multiple levels of prototypes that have to be tested in various labs.
Documentation stage is very important for medical devices, and has to be done at every step of design control. One of these documents is the market requirement document. It includes all the results of the initial discussions and research with doctors.
Then, intellectual property (IP) strategy needs to be decided. And if one is planning to branch out to different countries, IP strategy for each of those countries needs to be planned as well.
Programme plan for the product is also important. The audience should chart out plans for their products around three years in advance.
The alpha and beta prototypes
Once all relevant data has been collected and analysed, prototypes are created. The first is the alpha prototype. This is meant only for testing, and not for the market. Electromagnetic interference and electromagnetic compatibility tests are also performed at this stage.
Along with the design of the alpha prototype, there are certain regulatory and clinical considerations that need to be kept in mind. There are 14 such notified devices, and Drug Controller General of India (DCGI) needs to be informed about any products related to those devices. Only after approval has been granted, clinical trials can be conducted. Some of these devices are intraocular lenses, orthopaedic implants, heart valves and cardiac stents.
Getting through regulations
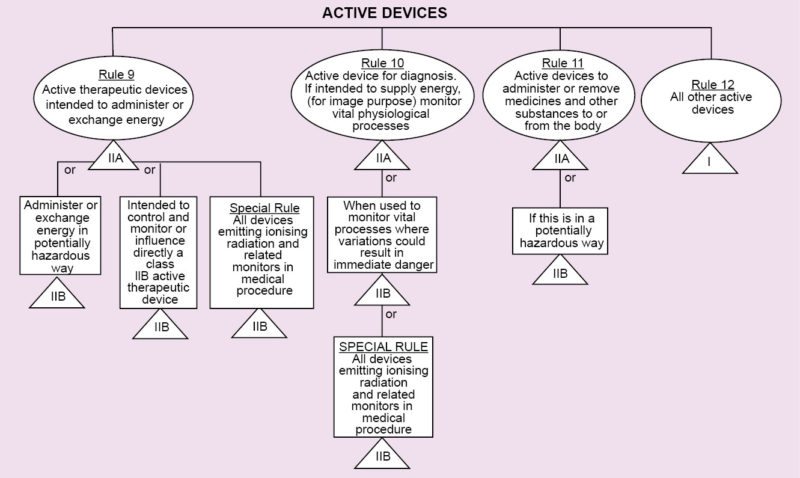
Regulations vary from country to country. In Europe, regarding medical devices, there is CE MDD. CE refers to European Conformity standards for products, and MDD (Medical Devices Directive) refers to standards that are specifically meant for medical products. It deals with three classes of devices that have been classified depending on the amount of risk that the device poses to the patient.
Class I refers to normal medical products that can be disposed of, such as surgical dressings and stethoscopes.
Class II is divided into two categories, A and B. Class IIA consists of medium-risk devices like tracheal tubes. Class IIB consists of higher-risk devices that are surgically-invasive or implantable like intraocular lenses.
Class III devices are the highest-risk implantable devices such as stents and radiology equipment. Based on the results of the tests conducted during the previous stage, the beta prototype is created, which is one step closer to the market.
The USA has certain Food and Drug Administration regulations that involve a similar classification method as CE MDD. It has a category known as 510(k). This is for products that have been developed that are similar to existing devices. The products may have different engineering designs, but these have the same clinical outcomes, so no clinical trials and pre-market surveys are needed.
Essentially, a company needs to show that their device is similar to an existing device, along with the lab test reports of the existing product. Regulatory authorities also provide a detailed framework of rules and regulations, and one should follow the regulations that are applicable to the product. Clinical literature is also important.

Clinical engagement
Another important factor that has to be kept in mind is clinical engagement. This can be done in two ways, either with clinical trials or with clinical studies. Clinical trials are large-scale, but in the case of clinical studies very few people are involved. A clear-cut plan is required for clinical engagement, and the plan needs to be approved by the DCGI as well as hospital committees.
Designing in India is different
Ultimately, it is important to keep in mind that designing medical devices in India is a completely different process compared to the rest of the world. In India, the market is doctor-driven. Even though consumers are the real users of all medical devices, they are not the final decision makers. Doctors are the ones who decide which products are going to be used.
There is also the issue of varying rules and regulations for each state in India.
Last, the whole system is more oriented towards treatment rather than prevention. This is unfortunate, because the cost of treatment is usually more than the cost of prevention.
Hopefully, we will see more entrepreneurs taking the road to innovate for the medical sector.
Nitesh Kumar Jangir is co-founder, Coeo Labs Pvt Ltd. The article has been curated by Ahalya Mandana, freelance writer for EFY





